The amino acid section on the Organic Acid Test (OAT), which includes Markers 62-75, often generates questions for practitioners new to the OAT. These metabolic metabolites are typically used by neonatologists, pediatricians, and geneticists to identify inborn errors of metabolism, so they are not necessarily common measurements. Inborn errors of metabolism are enzymatic defects in the metabolism of fats, carbohydrates, proteins, and/or the urea cycle, pyruvate or citric acid cycle, or the electron transport chain. For genetic abnormalities to occur on this part of the test, the elevations must be significantly elevated (meaning well above the second standard deviation). Mild elevations may have some importance, depending on the metabolite being measured, which is denoted in the generated interpretation, and low values rule out these genetic abnormalities.
To explain the metabolites in this section, below will be a brief overview of the measured metabolites, and the related genetic disorder, or other reasons associated with elevations.
Markers 62-66
2-Hydroxyisovaleric Acid, 2-Oxoisovaleric Acid, 3-Methyl-2-oxovaleric Acid, 2-Hydroxyisocaproic Acid, and 2-Oxoisocaproic Acid- These metabolites are related to the conversion of branched-chain amino acids (BCAA) into energy in skeletal muscles. Slight elevations may result from lactic acidosis, episodic ketosis, or deficiencies of the vitamins thiamine or lipoic acid. Extreme or multiple elevations can signify Maple Syrup Urine Disease (MSUD). This disorder is defined by a deficiency of an enzyme complex called branched-chain alpha-keto acid dehydrogenase, which is required to metabolize the BCAAs. Below is a figure to help visualize the pathway.
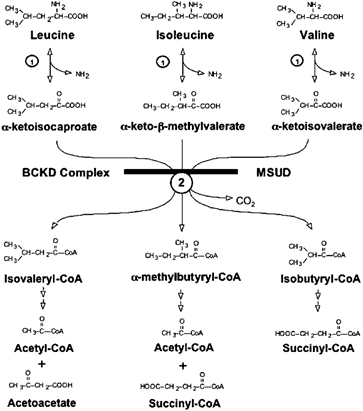
Figure 1: Oxidative degradation of the BCAAs leucine, isoleucine, and valine. The transamination of BCAA is catalyzed by a single branched-chain aminotransferase (reaction 1). The oxidative decarboxylation of BCKAs is catalyzed by the single mitochondrial branched chain a-ketoacid dehydrogenase complex (reaction 2). The metabolic block at the second reaction results in MSUD.
**Adapted from “Lessons from Genetic Disorders of Branched-Chain Amino Acid Metabolism,” by Chuang DT, Chuang JL, and Wynn RM. 2006. Journal of Nutrition. 136. 243S-9S. 10.1093/jn/136.1.243S.Marker 67
2-Oxo-4-methiolbutyric Acid- This metabolite may be elevated in inborn errors of methionine metabolism; examples include: S-adenosylhomocysteine (SAH) hydrolase deficiency, methylenetetrahydrofolate reductase deficiency (MTHFRD), methionine adenosyltransferase deficiency, and glycine N-methyltransferase deficiency. If suspected, genetic testing is warranted via plasma amino acids.
Markers 68-70
Mandelic Acid, Phenyllactic Acid, and Phenylpyruvic Acid- These metabolites are associated with the genetic disorder phenylketonuria (PKU), which is caused by a deficiency in the cofactor tetrahydrobiopterin (or BH4). In cases where very mild elevations occur, it may be caused by excessive intake of phenylalanine (dietary or supplementation). Insufficiencies or deficiencies of biopterin may be another potential. Figure 2 is a visualization of phenylalanine and tyrosine breakdown as it relates to PKU and biopterin defects.
Mandelic is also associated with PKU though not present on the figure, and not as well understood. It is believed to be formed from phenylpyruvic acid directly in large amounts. It is a marker that can also potentially reveal an exposure to styrene, measured on the GPL TOX test, when elevated, since it is generated in the metabolism of styrene.
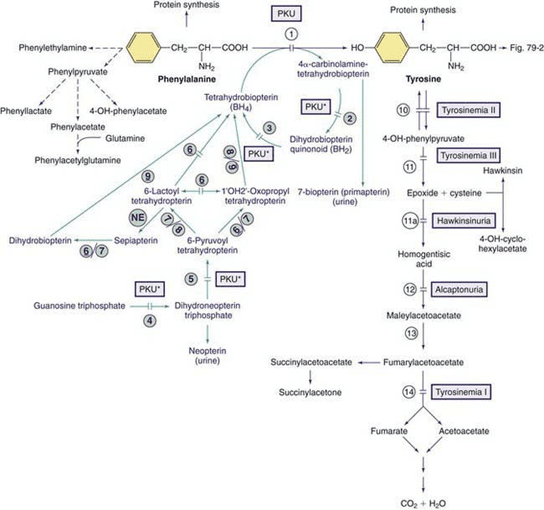
Figure 2: Pathways of phenylalanine and tyrosine metabolism. Enzyme defects causing genetic conditions are depicted as horizontal bars crossing the reaction arrow(s). Pathways for synthesis of cofactor BH4 are shown in purple. PKU* refers to defects of BH4 metabolism that affect the phenylalanine, tyrosine, and tryptophan hydroxylase.
*Adapted from Defects in Metabolism of Amino Acids. Phenylalanine. 2015.Marker 71
Homogentisic Acid – Homogentisic acid is elevated in the genetic disorder homogentisic aciduria (also known as, alkaptonuria). Homogentisic acid damages cartilage and heart valves, with the potential to precipitate kidney stones and stones in other organs. Symptoms typically occur after 30 years of age, but dark urine (almost black) is apparent at birth. Slight increases may indicate the heterozygous genetic carrier state of the disease.
Marker 72
4-Hydroxyphenyllactic Acid – Increased values are commonly associated with tyrosinemias, which can result from immature development of enzyme synthesis in infants or genetic deficiencies. Slight increases may be due to increased tyrosine intake, bacterial gut metabolism (potentially clostridia species), short bowel syndrome, or liver disease.
Marker 73
N-Acetylaspartic Acid – Elevated N-acetylaspartic acid is due to the genetic disorder Carnavan’s disease, a potentially fatal disease-causing spongy degeneration of the brain.
Marker 74
Malonic Acid – Associated with the genetic disorder malonyl-CoA decarboxylase deficiency or malonic aciduria with normal malonyl-CoA decarboxylase activity. Slightly elevated values in urine are unlikely to be clinically significant.
Marker 75
4-Hydroxybutyric Acid- Very high levels may indicate the genetic disorder involving succinic semialdehyde dehydrogenase deficiency. A decrease in this enzyme leads to an increase in GABA and GHB, especially in the brain and spinal cord, leading to a myriad of neurological disorders.
In conclusion, these metabolites are measuring the functionality of enzymes associated with the body’s utilization of certain amino acid groups, going beyond digestion and absorption of amino acids. Therefore, low values do not indicate defects in breaking down or absorbing protein, rather the inference is the associated enzyme is functioning well because there is no backup of that metabolite. Due to the rarity of these genetic diseases, it is uncommon to see elevations in this section, rather, it is more common to observe low values. With that in mind, when elevations in this section do occur, it may be helpful to utilize the consultations services to help further investigate the potential need for genetic testing.
References
1. Rare Disease Database. Maple Syrup Urine Disease. 2020. SOURCE
2. .Chuang, David & Chuang, Jacinta & Wynn, R.. (2006). Lessons from Genetic Disorders of Branched-Chain Amino Acid Metabolism. The Journal of nutrition. 136. 243S-9S. 10.1093/jn/136.1.243S.
3. Alfonso, I. Neuroneonatology. Errors of metabolism; pg 72. Accessed 2/25/2021 SOURCE
4. Rezvani I and Melvin JJ. Defects in Metabolism of Amino Acids. Phenylalanine. 2015. SOURCE
5. Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis. 2009 Jun;32(3):333-42. doi: 10.1007/s10545-009-1067-2. Epub 2009 Feb 9. Erratum in: J Inherit Metab Dis. 2009 Jun;32(3):457. PMID: 19234759.
6. Shintaku H. [Biopterin and child neurologic disease]. No To Hattatsu. 2009 Jan;41(1):5-10. Japanese. PMID: 19172809.
7. Godwin BL, Ruthven CR, Sandler M. Phenylpyruvic acid may be a direct precursor of mandelic acid without intermediate transamination of phenylalanine. Biochem Pharmacol. 1993 Dec 3;46(11):2109-10. doi: 10.1016/0006-2952(93)90656-h. PMID: 8267662.
8. Chuang, David & Chuang, Jacinta & Wynn, R.. (2006). Lessons from Genetic Disorders of Branched-Chain Amino Acid Metabolism. The Journal of nutrition. 136. 243S-9S. 10.1093/jn/136.1.243S.
9. Elia VJ, Anderson LA, Macdonald TJ, Carson A, Buncher CR, Brooks SM. Determination of urinary mandelic and phenylglyoxylic acids in styrene exposed workers and a control population. Am Ind Hyg Assoc J. 1980 Dec;41(12):922-6. doi: 10.1080/15298668091425879. PMID: 7468463.
10. Medline Plus and Genetic Home Reference. Succinic semialdehyde dehydrogenase deficiency. 2020. SOURCE



